



Gaussian量化模擬:實例操作之自旋多重度的判斷,在修改Gaussian輸入文件時,自旋多重度往往是困擾入門者的難題,換言之,只要能夠知道模型分子的電子占據情況,即可算出該體系的自旋多重度,簡單體系,對于比較簡單的體系,可根據原子或分子軌道理論來判斷,根據計算所得的靜電勢,可進一步結合VMD軟件進行處理,還可以得到精美的分子靜電勢分布圖。
原子間的連接方式,這些字樣代表原子之間的連接關系,對于量化模擬毫無意義,故而可直接刪掉,這是由于量化模擬會在計算過程中根據原子各自電子云間的交疊情況自主判斷成鍵形式,因此軟件并不關心輸入模型中的化學鍵,換言之,在建模時,水分子中O和H之間畫成單鍵抑或三鍵,對于計算結果沒有任何影響,那么此時,Results欄中的“Vibrations”可選,從中可以分析分子的所有振動形式及對應的紅外光譜出峰位置,進而生成該分子的理論紅外光譜。
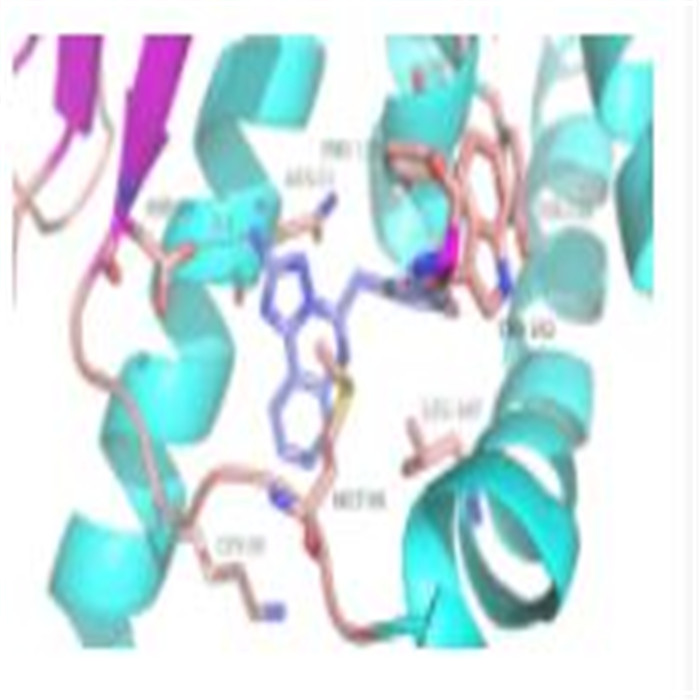
輸入文件:文件后綴名通常為,gjf,包含模擬任務的計算資源分配(核數、內存使用情況)、計算方法和精度、任務要求及計算模型等信息,輸出文件:文件后綴名通常為,out或,log,除輸出作為計算結果的結構模型、軌道、密度矩陣、電荷布局等信息外,還包括了部分計算過程中輸出信息,對于大多數含有過渡金屬的結構、自由基結構、激發態,如二茂鐵、基態氧分子、羥基自由基等,均為開殼層體系,自旋多重度大于1,需做進一步判斷。